



By Sophie Arthur
July 21, 2020
Time to read: 4 minutes
Our bodies are made up of 37.2 trillion cells, according to our latest estimates. Within this beautiful mixture, there are thousands, probably even millions, of different types of cell. But despite this diversity of cell types, each still shares the same DNA. There are many different ways in which a given type of cell will ensure that the correct genes are switched on for it to be able to properly function, including how its DNA is packaged.
In reality, very little of our DNA actually codes for genes, which in turn create proteins. In fact, most of our DNA is made up of so-called “non-coding” regions. These regions can act as regulators instead, helping to fine tune which genes are switched on in a given cell. Enhancers are one type of these molecular “switches”.
Surprisingly, enhancers can often be found a long way away on the DNA thread from the gene which they help to switch on or off. However, our DNA is not stored in the cell’s nucleus as one long, linear string – there would be no room for the nearly two metres of it if that were the case. Rather, it is tightly packaged in the nucleus. Beyond space-saving, this packaging allows enhancers to reach the genes they control by “looping over” towards them in 3D. The way in which DNA is packaged in the nucleus of a cell is therefore important in determining which genes are switched on. How exactly enhancers form these loops is not completely understood.
A prime suspect in this mysterious process is a protein complex called cohesin. Cohesin forms a “protein ring” that entraps DNA, which is important for holding two copies of the same DNA, known as “sister chromatids”, together so they are distributed correctly when cells divide. However, cohesin can also entrap two parts of the same DNA molecule, forming loops. The key question, therefore, is whether cohesin is the main driving force for keeping enhancers and their “target” genes together.
Research from the Functional Gene Control group at the MRC LMS, published on 21 July in Cell Reports, in collaboration with researchers at the Institute of Molecular Pathology in Vienna and the Babraham Institute in Cambridge, sheds light on this question. Researchers degraded cohesin very rapidly in cells and used a high-resolution biochemical technique called Capture Hi-C to explore what would happen to the chromosomal contacts between enhancers and their target genes when this pivotal architectural protein was absent.
They confirmed that many such contacts were indeed disrupted when cohesin was no longer present. This could explain why some genes regulated by these enhancers also inappropriately switched on or off. However, more surprisingly, they found that many other enhancers still remained connected to their target genes despite cohesin being completely degraded, and thus the activity of these genes also remained unchanged.
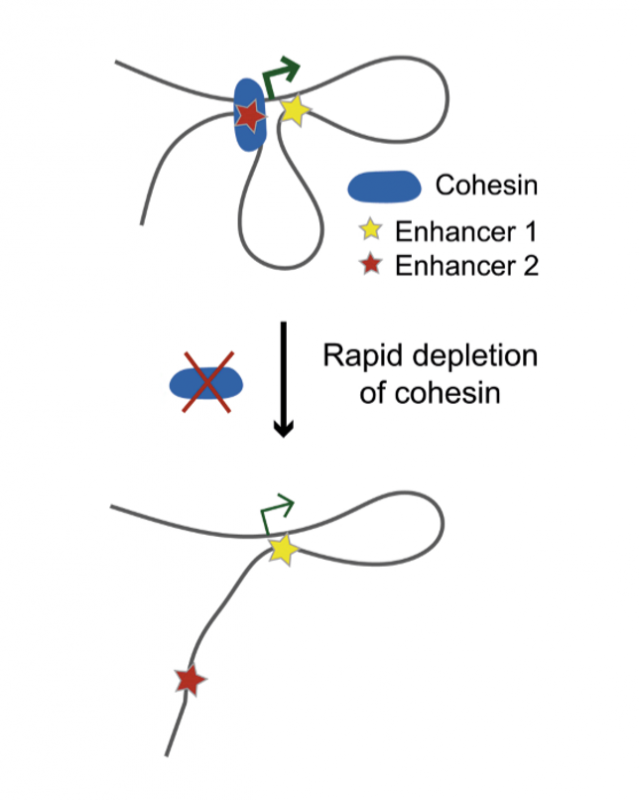
The researchers then employed machine learning techniques to detect the specific characteristics of those enhancer-gene contacts that required cohesin, and those that did not. They found that the contacts that required cohesin tended to span longer distances, and that the enhancers participating in these contacts were commonly localised to specific parts of the genome known as topologically associated domain (TAD) boundaries. TAD boundaries are a feature of genome organisation which is highly conserved across species. In contrast, cohesin-independent contacts typically spanned shorter distances. Enhancers participating in these contacts were less restricted in their genomic location.
Mikhail Spivakov, Head of the Functional Gene Control group at the LMS and a senior author on this study, discussed the implications of these findings:
“These results confirm that this important architectural protein, cohesin, links enhancers and their target genes together, but suggest that this is not the whole story. This can explain why many genes seem highly resilient to cohesin removal, something that has puzzled researchers for years. Our findings will pave the way to predicting which genes may be deregulated when cohesin is not functioning properly, for example, when it is mutated in cancers. They also prompt a quest for proteins other than cohesin that can facilitate contacts between enhancers and the genes they control”.
‘Cohesin-dependent and independent mechanisms mediate chromosomal contacts between promoters and enhancers’ was published in 21 July in Cell Reports. Read the full article here.