



By Sophie Arthur
June 26, 2020
Time to read: 4 minutes
The human genome is made up of approximately 3 billion bases. Amongst many other things, all these bases code for every single one of our genes. These genes are made up of just 4 different letters; A, T, C and G. Our DNA can get surprisingly far just using different combinations of these 4 letters. But there are other ways in which these bases can go that bit further by using so-called epigenetic modifications.
There are various different types of epigenetic modification, but one of the most prominent is methylation. These additions act like tiny chemical flag on top of certain bases in the genetic code. These flags tell the cellular machinery to change the structure of the DNA, or even determine whether it is read or not. In animals, methylation occurs most often on the C base, which is short for cytosine. Cytosine methylation is an ancient epigenetic modification. Yet its function and the number of times it appears in genomes is hugely variable.
Research, led by our Epigenetic Inheritance and Evolution group at the MRC LMS in collaboration with Frank Jiggins lab at University of Cambridge, has characterised the evolution of cytosine methylation across arthropods. Arthropods are a group of animals that are invertebrates with an exoskeleton, a segmented body and jointed legs. This includes crabs, centipedes, fruit flies, mites and more.
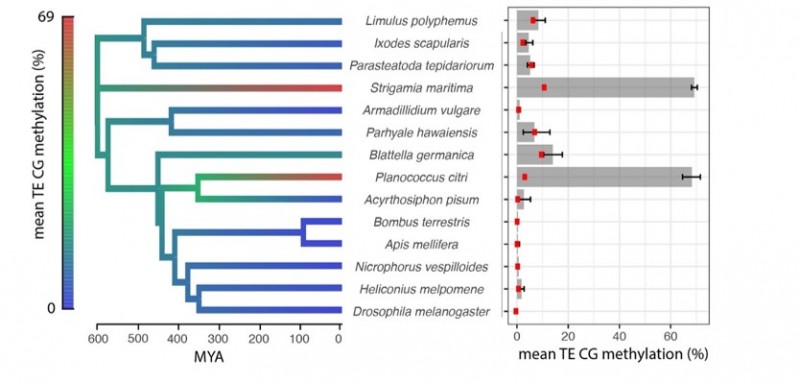
The researchers looked at horseshoe crabs, butterflies, mealybugs and centipedes. They analysed the patterns of methylation across their genes, and how those chemical flags affected the activity of those genes too. This study starts to trace how methylation patterns have changed across arthropod evolution.
The common ancestor of all these arthropods had low levels of methylation, but much higher levels had evolved independently in both centipedes and mealybugs. Patterns in the positioning of these methylation tags were observed. These modifications seemed to happen in similar sets of genes across all the creatures. These observations might help explain how this type of epigenetic modification pattern is put in place in other species too, including humans.
Peter Sarkies, Head of the Epigenetic Inheritance and Evolution group at the LMS and senior author on this study discussed the paper further:
“Despite these ancient, most conserved features, the most exciting thing was how rapidly different patterns seemed to emerge in a few isolated examples. In both the centipede and the mealybug, we saw much higher levels of methylation. In fact, their methylation was much more like mammalian methylation – we found it within almost all so-called ‘jumping genes’ and also it was being used to shut down some genes. Amazingly, these patterns had emerged separately in these two organisms over a short timescale.”
Peter further discussed what this means for next steps of the research:
“Although speculative, I’d like to suggest that methylation as we found in the original arthropod was probably the most ancient type of methylation in animals. But for some curious reason, methylation is always poised to rapidly target a much higher proportion of selfish DNA, perhaps following invasion of a particular new invading sequence. Once this happens, it naturally acquires the ability to shut down genes and this can be used to regulate cell identity, just as happens in mammals and a few creepy crawlies today. We’ll have to study many more different groups of animals to really test this idea though – so if anyone fancies collecting a diverse group of molluscs or jellyfish we’d love to hear from you.”
‘Widespread conservation and lineage-specific diversification of genome-wide DNA methylation patterns across arthropods’ was published in PLoS Genetics on 25 June. Read the full article here.