



By Helen Figueira
August 27, 2010
Time to read: 5 minutes
 Lipoprotein Group finds new mechanism in atherosclerosis
Lipoprotein Group finds new mechanism in atherosclerosis
‘Bad’ cholesterol – that bane of cream-cake lovers’ lives – is one of the leading factors contributing to atherosclerosis, the blocking of arteries by fatty substances that can eventually lead to heart attack or stroke. For a number of years, a model of physiological cholesterol regulation has been developed that has focused solely on the degradation of the receptor LDLR, because loss of this receptor is linked to increased blood cholesterol (hypercholesterolemia) and atherosclerosis.
Recently, a paper by members of the CSC’s Lipoprotein group has challenged this assumption, and found that the reality is more complex than has been assumed. The paper has also been highlighted in an Editorial piece for the publishing journal, Arteriosclerosis, Thrombosis and Vascular Biology.
Like a great many questions in biology, the starting point for understanding the normal metabolism of cholesterol lies in cases of extremes. Familial Hypercholesterolemia (FH) is a condition characterised by increased levels of bad cholesterol and a greater risk of premature coronary heart disease. It is caused by a mutation, either of LDLR (low density lipoprotein receptors – see ‘Additional Background’, below) which mop up LDL (bad cholesterol); or of apolipoprotein B100 (apoB100), the protein component of LDL that interacts with LDLR. In 2003, the occurrence of a mutated serine protease D374Y-PCSK9 was associated with FH. The mutation causes an increased degradation of LDLR, leaving fewer receptors to mop up LDL and increasing the arterial plaques that constitute atherosclerosis. In 2005, at the opposite end of the ‘cholesterol spectrum’ from FH, a mutation of PCSK9 found in 2% of African Americans that reduced its function was linked to an 88% decrease in coronary heart disease risk. It is unsurprising that a great deal of clinical research has focused on PCSK9 and more specifically on its regulation of cholesterol levels through LDLR, but was that the whole story?
In order to test whether LDLR degradation was the sole mechanism of LDL homeostasis, the Group led by Anne Soutar took mice expressing normal – or ‘wildtype’ – PCSK9 and the D374Y version identified in FH, plus a group of control mice not expressing PCSK9. They found that levels of cholesterol in the blood for the D374Y mice was highest, followed by wildtype PCSK9, with control mice having the lowest levels; the level in D374Y was further exacerbated by a cholesterol-rich diet.
Our diet is only part of the problem: our risk of developing atherosclerosis is also influenced by our genes. A complex cascade of regulatory feedback loops is subject to the programming in our genetic code, making individual physiology as unique as personality.
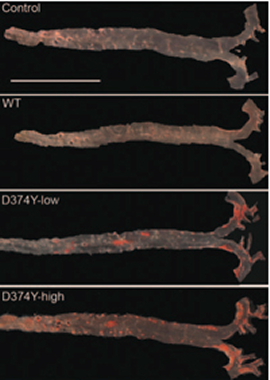
Above: Aortic lesions observed in D374Y Mice
As anticipated in light of results from previous studies, levels of LDLR in the D374Y and wildtype PCSK9 were lower than in control mice. However, it was also found that secretion of apoB-containing lipoproteins by liver cells was increased in the D374Y mice, and that blood plasma samples taken from these mice during fasting contained much larger lipoproteins, termed Very-Low Density Lipoproteins (VLDL). This is important because such ‘triglyceride-rich’ lipoproteins, alongside increased levels of apoB, are seen in human FH patients. Larger lipoproteins are associated with increased deposition of the atheroma plaques that constitute atherosclerosis.
The findings of the CSC group show that reduced LDLR isn’t the sole mechanism behind hypercholesterolemia, and it is hoped that further studies of D374Y mice may lead to new effective therapies to reduce atherosclerosis.
SJ
Cholesterol itself is vital in maintaining permeability in cell membranes and is an important metabolite for many physiological functions, including bile production. Transported around the body in lipoprotein vesicles of various size, it is those vesicles termed ‘low density lipoproteins’ (LDL) that constitute ‘bad’ cholesterol. LDL vesicles comprise a greater proportional mass of cholesterol than ‘High Density Lipoprotein’ (HDL) vesicles – so-called ‘good’ cholesterol – which are about half the physical size of LDLs. LDL vesicles are mopped up by an LDL receptor (LDLR), but the level of LDLR is kept in check by PCSK9; an example of the multiple levels of feedback involved in mammalian metabolism. apoB stabilises and solubilises the cholesterol-carrying lipoprotein vesicles as they are transported in blood plasma, much as detergents solublise oil droplets in washing-up water. Increased apoB such as observed in the D374Y mice and human FH allows the formation of larger lipoprotein vesicles, which increases the risk of the plaques characteristic of atherosclerosis.
This work appears in Atherosclerosis, Thrombosis and Vascular Biology (link to article)
Herbert, B., Patel, D., Waddington, S. N., Eden, E. R., McAleenan, A., Sun, X.-M., Soutar, A. K., July 2010. Increased secretion of lipoproteins in transgenic mice expressing human d374y pcsk9 under physiological genetic control. Arterioscler Thromb Vasc Biol 30, 1333-1339.
The Editorial, Beyond LDL Cholesterol, a New Role for PCSK9 by ON Akram et al. is available here
Our diet is only part of the problem: our risk of developing atherosclerosis is also influenced by our genes. A complex cascade of regulatory feedback loops is subject to the programming in our genetc code, making individual physiology as unique as personality.
At above: Aortic lesions observed in D374Y Mice
Our diet is only part of the problem: our risk of developing atherosclerosis is also influenced by our genes. A complex cascade of regulatory feedback loops is subject to the programming in our genetc code, making individual physiology as unique as personality.
At above: Aortic lesions observed in D374Y Mice