



By Jenna Stevens-Smith
July 12, 2018
Time to read: 4 minutes
Identifying which mutations and which genes drive cancer is difficult, especially for mutations that are rare or mutations between genes where it is unclear which genes are being affected. Gathering more data from more samples improves the odds of identifying new genes and hence new therapeutic targets, but generating this data is painstaking and expensive work. In new research published in Nature Communications, the largest scale study of its kind to date, researchers at the MRC LMS at Imperial College London explored how sequencing tumour DNAs to a much greater depth can add confidence to our knowledge of which genes are driving disease.
When a single cell accumulates cancer causing mutations, it becomes more likely to divide and eventually form a tumour. By examining the full spectrum of mutations present in cells that have not yet formed tumours and comparing them to mutations at different stages of tumour development the researchers were able to better discriminate between mutations that drive disease from those that are harmless bystander mutations. Typically, most studies of cancer mutations only study the “clonal” mutations that are present in the majority of the tumour. By using the murine leukaemia virus (MuLV)-induced lymphoma model, the researchers were able to additionally study the vast numbers of mutations that are present in only a few cells in both lymphomas and non-malignant tissue.
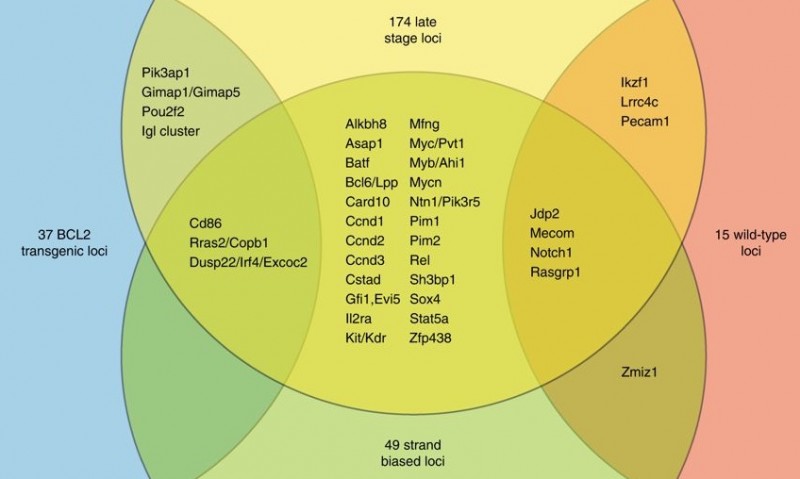
In total 700,000 mutations were identified from more than 500 malignancies collected at timepoints throughout tumour development, only 3,000 of which were the highly abundant clonal mutations. The abundance of subclonal mutations in this study allowed the researchers to dig deeper than prior studies, especially for mutations outside genes. By examining clonal and subclonal mutation frequencies at different timepoints throughout tumour development, the study identified hundreds of genes that contribute to disease that could not have been identified through the sole use of clonal mutations.
The density of the data generated allows construction of an online genome-wide map of hundreds of genomic regions that contribute to tumour development. By comparing these regions to human cancer genome datasets, they found overlap with more than 100 known genes and regions implicated in non-Hodgkin lymphoma. Several hundred additional regions were also implicated suggesting further study of these in human datasets is warranted.
The research combined interdisciplinary expertise from biological, medical and data science researchers from the MRC London Institute of Medical Sciences, Institute of Clinical Sciences, Imperial College London; Centre for Systems and Synthetic Biology, Department of Computer Science, Royal Holloway, University of London; in collaboration with Imperial College Healthcare NHS Trust and Centre for Pathology/Haematology, Imperial College London.
Discussing potential next steps for the research, senior author Dr Anthony Uren said:
“The research has highlighted a number of promising targets that warrant further investigation, particularly modulators of anti-tumour immune response.”
The researchers have developed an online resource that can be used by fellow researchers to prioritise rare mutations from human tumours for further study: http://mulv.lms.mrc.ac.uk.
Subclonal mutation selection in mouse lymphomagenesis identifies known cancer loci and suggests novel candidates by Philip Webster, Joanna C. Dawes and Anthony G. Uren, et al is published in the journal Nature Communications (https://doi.org/10.1038/s41467-018-05069-9).