



By Sophie Arthur
January 30, 2020
Time to read: 3 minutes
Telomeres consist of repeats of a DNA sequence that cap off the end of chromosomes and prevent them from being recognised as a break in our DNA. Think of them like the cap at the end of your shoelaces which stops them from unravelling and fraying. Because of the way our DNA copies itself, after every replication event, the ends of the chromosomes are shortened a little. Having telomeres at the end of our chromosomes to act as buffers means that we don’t lose precious genetic material that could code for essential proteins. Eventually, telomeres will become too short to do their job, causing our cells to age and stop functioning properly.
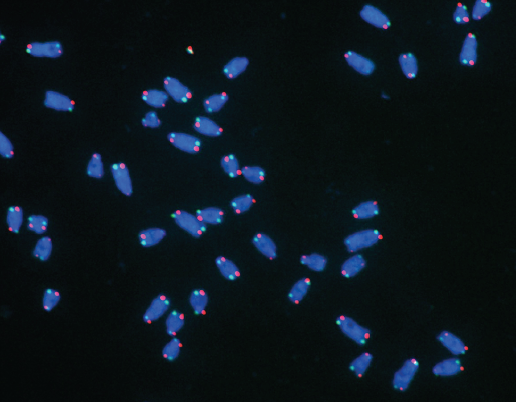
However, cancer cells can counteract this issue of shortening telomeres to become immortal, and there are two main mechanisms behind how they do that. The first way is common to about 90% of cancers and involves the reactivation of the enzyme telomerase – which works to maintain the length of telomeres by adding more of those DNA sequence repeats to the ends of chromosomes. However, some cells can be telomerase-negative and so use an enzyme-independent mechanism called Alternative Lengthening of Telomeres (ALT). Relatively little is known about ALT, but it involves extending telomeres by using the DNA sequence repeats that are found in telomeres as a template. But how cells decided to use this ALT mechanism was not known.
Research from our Telomere Replication and Stability group published in the journal eLife on 14 January describes how the protein TRF1 acts as a protective cap and stops the telomeres being recognised as a DNA break. But this comes with its own challenges, as this protective structure makes it difficult for the molecular machinery the replicates our DNA to pass causing a lot of replication stress. TRF1, however, was previously known to ease this replication stress, and there was also a link between replication stress and this mechanism of ALT activation – but how the three all linked together was not understood fully.
Rosa Maria Porreca, first author on the paper, discussed what they reported in this paper:
“We used a proteomics approach to reveal that the telomeres in cells without the protein TRF1 were associated with higher numbers of proteins linked to the DNA damage response and recombination. This means that cells without the protein TRF1 closely resemble that of ALT telomeres, which are characterised by the presence of these DNA damage proteins as the telomeres are being recognised as a break in the DNA – exactly what the telomeres and TRF1 are trying to prevent.”
This research reports that TRF1 has a critical role for preventing cancer cells extending their telomeres using the ALT mechanism, and as such will stop them becoming immortal. The next steps for the team are to explore more of what factors are involving in the initiation of this ALT mechanism which could provide another potential alternative for treating cancer cells.
This paper is also a celebration for Jean-Baptiste Vannier, Head of the Telomere Replication and Stability group, and the rest of his team as it is the first publication from the lab.
‘TRF1 averts chromatin remodelling, recombination and replication dependent-break induced replication at mouse telomeres’ was published in eLife on 14 January. Read the article here.