



By Helen Figueira
June 1, 2010
Time to read: 5 minutes
 New detection of subtle expression
New detection of subtle expression
Exactly how does the interaction of genes give rise to a particular phenotype at the level of the whole organism? It is a problem that would be intractable without powerful computational analyses: over the last few decades, the processor speed and storage capacity obtainable for a given price has increased geometrically, putting solutions to big problems within reach of an ever greater number of researchers. But raw silicon statistics aren’t enough. True computational power in bioinformatics is afforded by employing the right statistical tools: elucidating meaning in vast genomic data sets that may contain many blind alleys and dead ends – red herrings, and elusive treasures.
A relatively new but standard approach in transcriptional profiling is to identify a sample of probe gene sets whose expression is under genetic control (‘Expression Quantitative Trait Loci’ – eQTLs) and to look at how variation of these affects a particular phenotype. Yet analyses across tissues have previously been carried out by comparing results from multiple, single-tissue experiments – an approach that, due to variable accuracy rates (false positives and negatives) across tissues, leads to inflated discrepancies in identification of common eQTL lists. Application of standard univariate analytical methods further exacerbate the already-existing detection bias towards cis-eQTLs – which are more highly expressed, caused by genomic sequence variants residing within or close to the gene itself – at the expense of trans-eQTLs, which are caused by genetic variations distal from the gene itself. The more subtle effects of the latter are effectively sacrificed in analyses designed to clarify statistical significance in highly dimensional data.
New detection of subtle expression
Exactly how does the interaction of genes give rise to a particular phenotype at the level of the whole organism? It is a problem that would be intractable without powerful computational analyses: over the last few decades, the processor speed and storage capacity obtainable for a given price has increased geometrically, putting solutions to big problems within reach of an ever greater number of researchers. But raw silicon statistics aren’t enough. True computational power in bioinformatics is afforded by employing the right statistical tools: elucidating meaning in vast genomic data sets that may contain many blind alleys and dead ends – red herrings, and elusive treasures.
A relatively new but standard approach in transcriptional profiling is to identify a sample of probe gene sets whose expression is under genetic control (‘Expression Quantitative Trait Loci’ – eQTLs) and to look at how variation of these affects a particular phenotype. Yet analyses across tissues have previously been carried out by comparing results from multiple, single-tissue experiments – an approach that, due to variable accuracy rates (false positives and negatives) across tissues, leads to inflated discrepancies in identification of common eQTL lists. Application of standard univariate analytical methods further exacerbate the already-existing detection bias towards cis-eQTLs – which are more highly expressed, caused by genomic sequence variants residing within or close to the gene itself – at the expense of trans-eQTLs, which are caused by genetic variations distal from the gene itself. The more subtle effects of the latter are effectively sacrificed in analyses designed to clarify statistical significance in highly dimensional data.
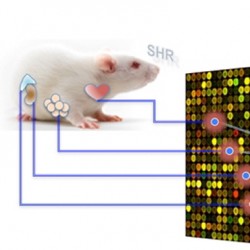
But now, CSC Integrative Genomics and Medicine Group Head Enrico Petretto and coworkers at Imperial College department of Epidemiology and Biostatistics (Prof. Sylvia Richardson) have developed a new fully multivariate Bayesian approach to eQTL analysis in single and jointly across multiple tissues (PLoS Comput Biol 6, e1000737, 2010). The team looked at set of representative genes across four tissues (fat, kidney, adrenal and heart) in a panel of recombinant inbred strains derived by cross-breeding the Spontaneously Hypertensive and Brown Norway rats. Using the new Bayesian approach they detected many cis and trans-eQTLs – verified experimentally – that would not have been detected by other methods, and were able to disentangle tissue specific genetic regulation of transcription from systems-level effects. Critically, a more than fivefold eQTL detection rate increase from the joint analysis of the four tissues was seen as compared to that determined by comparing single-tissue analyses.
The work of the group is a big step forward in terms of looking at regulation of gene expression at the whole organism level. It is also a highly flexible approach, and is of great importance to understanding the gene expression changes underlying complex phenotypes in humans. SJ
But now, CSC Integrative Genomics and Medicine Group Head Enrico Petretto and coworkers at Imperial College department of Epidemiology and Biostatistics (Prof. Sylvia Richardson) have developed a new fully multivariate Bayesian approach to eQTL analysis in single and jointly across multiple tissues (PLoS Comput Biol 6, e1000737, 2010). The team looked at set of representative genes across four tissues (fat, kidney, adrenal and heart) in a panel of recombinant inbred strains derived by cross-breeding the Spontaneously Hypertensive and Brown Norway rats. Using the new Bayesian approach they detected many cis and trans-eQTLs – verified experimentally – that would not have been detected by other methods, and were able to disentangle tissue specific genetic regulation of transcription from systems-level effects. Critically, a more than fivefold eQTL detection rate increase from the joint analysis of the four tissues was seen as compared to that determined by comparing single-tissue analyses.
The work of the group is a big step forward in terms of looking at regulation of gene expression at the whole organism level. It is also a highly flexible approach, and is of great importance to understanding the gene expression changes underlying complex phenotypes in humans. SJ
This work was recently highlighted in Nature Review Genetics (Research Highlights June 2010 11:6).