



By Katy G Pallister
June 8, 2021
Time to read: 3 minutes
By Katy Pallister
Beta cells are insulin-producing cells found in pancreatic islets – clusters of hormone-producing cells in the pancreas. When stressed by social, environmental, and metabolic factors, the beta cells become less specialised (dedifferentiate), and lose their ability to produce insulin and control circulating sugar levels – a process now thought to underpin diabetes.
Researchers from the Cellular Identity and Metabolism group at the MRC London Institute of Medical Sciences (LMS) have investigated beta-cell dedifferentiation in diabetes, and have uncovered how this loss of cell identity leads to the formation of permanent fibrous islet tissue and impaired function of the pancreas in diabetes.
“Historically diabetes was associated with beta-cell apoptosis (cell death) and dysfunction,” senior author, Dr Mathieu Latreille, Head of the Cellular Identity and Metabolism group at the LMS, said. “More recently, dedifferentiation of beta cells has also been revealed as a mechanism underlying a decrease in circulating insulin levels in diabetes. In this study we used a diabetic phenotype model to try to understand what other genes are dysregulated in response to dedifferentiated beta cells and how this impacts the pancreatic islets’ microenvironment.”
Using bulk and single-cell RNA sequencing, the team found that a beta-cell specific transcription factor called PDX1 was downregulated as part of the dedifferentiation process. PDX1 is a master regulator for beta cells that activates genes involved in insulin transcription and glucose sensing. However, the researchers discovered that PDX1 also binds and activates the gene Ovol2, which is enriched in epithelial cells. These are cells which line most internal and external surfaces of the body and its organs.
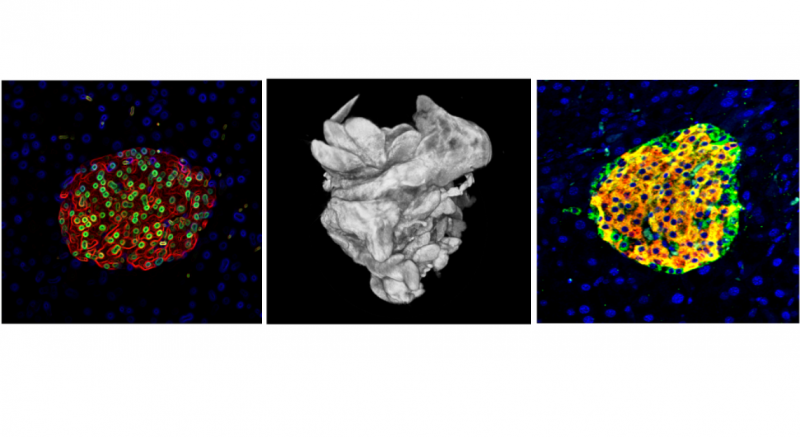 Although it remains unknown why beta cells, which are non-epithelial, contain epithelial-specific markers, the downregulation of PDX1 subsequently results in the downregulation of Ovol2 in the cells. With the loss of the epithelial marker, the beta-cell is reprogrammed into a mesenchymal cell, in a process called epithelial mesenchymal transition (EMT).
Although it remains unknown why beta cells, which are non-epithelial, contain epithelial-specific markers, the downregulation of PDX1 subsequently results in the downregulation of Ovol2 in the cells. With the loss of the epithelial marker, the beta-cell is reprogrammed into a mesenchymal cell, in a process called epithelial mesenchymal transition (EMT).
“In non-epithelial cells, such as insulin-producing beta cells, EMT is activated following an injury and is involved in organ regeneration,” Latreille said. “However, long-term activation of EMT triggers tissue fibrosis which impairs the function of organs, in this case the pancreas. Currently, fibrosis can’t be cured as treatments only reduce symptoms and the progression of the disease.”
EMT is also a key pathway activated in cancer cells, following a gene mutation or a tissue injury. The EMT process mobilises cancer cells, allowing them to travel from one organ to another and establish secondary tumours. The existence of EMT in beta-cells could mean that cancer treatments are a viable option to treat islet fibrosis in diabetes.
“EMT inhibitors are usually used in cancer patients, but now, because we have linked an EMT process to the dedifferentiation of beta cells in diabetes, we could use these inhibitors or agents that can stimulate Ovol2 activity to possibly improve the beta-cell function of diabetic individuals, and prevent the triggering of pancreatic fibrosis,” Latreille said.
The team’s discovery also provided an answer to a previous quandary. In humans, mesenchymal markers were found to be upregulated in individuals with diabetes compared to non-diabetics. Prior to this study, no-one knew why this was the case, or what the consequence of this was. However, this research suggests that these mesenchymal markers may be linked directly to beta-cell dedifferentiation, and that their presence could ultimately contribute to fibrosis.
‘Dysregulation of the Pdx1/Ovol2/Zeb2 axis in dedifferentiated β-cells triggers the induction of genes associated with epithelial–mesenchymal transition in diabetes’ was published online on 12 May 2021 in the journal Molecular Metabolism. Read the full article here.