 CSC team investigates ‘burst’ mechanism in Parkinson’s
CSC team investigates ‘burst’ mechanism in Parkinson’s
CSC researchers investigating brain cell death in Parkinson’s disease recently reported findings that demonstrate a link to genes important in controlling the function of mitochondria – the powerhouses that drive cellular reactions. Their combined genetic and neurophysiological study illuminates a mechanism by which changes in neuronal structure caused by genetic mutations leads to dysfunction and finally deterioration. This mechanism suggests a potential neuroprotective therapy for patients with the early stages Parkinson’s disease. The work is published in the Journal of Neurophysiology.
In the 1980s, a group of individuals exposed to the toxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) developed a chronic form of Parkinson’s disease. The toxin was shown to inhibit mitochondrial complex 1 – an enzyme found in the mitochondria of neurons and other cells that has a key role in maintaining cellular metabolism. Fast-forward 20 years, and mitochondrial dysfunction is thought to be a major player in the development of Parkinson’s disease (PD). Despite this connection, it has remained unclear exactly how this dysfunction leads to loss of dopaminergic neurons in the substantia nigra pars compacta (SNC dopamine neurons) – the decimation of which is a key feature of PD.
Genetic studies of patients with a family history of PD have identified several mutations of genes affecting mitochondrial architecture and function. None of these genes are selectively expressed in SNC dopamine neurons, however, indicating that some other mechanism must cause SNC deterioration in PD patients. One theory that has gained weight in recent years is that SNC dopamine neurons’ unique electrophysiological properties may make them particularly susceptible to degeneration in PD, so the CSC team investigated whether removing genes associated with PD in mice affected these properties.
Like other neurons, those of the SNC dopaminergic system work by transmission of electrical impulses supplied by a flow of chemical ions (charged particles) into and out of the neuron. This flow is controlled by channels that traverse the membrane, which selectively allow ions of the right size and charge to pass through. SNC dopamine neurons rely heavily on ‘small conductance calcium (2+)-activated potassium channels’ (‘SK channels’) to regulate their firing activity. As their name suggests, these channels are activated by calcium ions inside the neuron and, on opening, allow potassium ions to flow out of the cell. The activity of these channels is directly involved in controlling how ‘excited’ a neuron is and, indirectly, they provide information on calcium signalling within the cell. Both calcium dysregulation and enhanced excitability are thought to make SNC neurons highly susceptible to degeneration. Thus, disruption of SK channels function may represent a new insight into the early changes that occur in SNC neurons in Parkinson’s disease
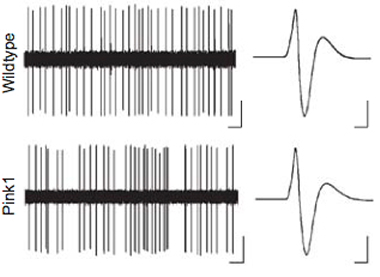
Out of a number of PD-associated genes that could affect excitability of SNC dopamine neurons, the team focused on PINK1 because it is almost exclusively active in mitochondria. By recording electrical traces of individual neurons, they compared SNC dopamine firing patterns of mice without PINK1 with those of normal mice. Mice missing PINK1 exhibited bursts of SNC dopamine neural activity punctuating a more irregular pattern; traces from the normal mice displayed greater regularity.
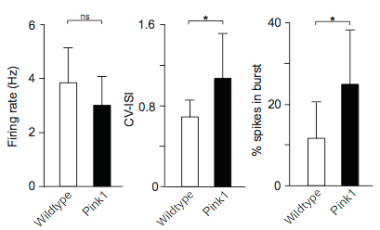
Bursts are thought to be important for dopamine signalling, but conversely it has also been suggested that they can increase intercellular calcium concentrations and stress in the cell.
Traces of electrical potentials from mice deficient in PINK1 suggested that SK channel function (see boxout, SNC channels) was impeded, suggesting that these may play an important role in the development of Parkinson’s disease. These channels are dependent on a supply of calcium ions to activate them, indicating that impaired calcium signalling is at fault. Rather than this being due to impairment of the channels that supply calcium ions, it was shown to result from a reduction in the number calcium ion release from the endoplasmic reticulum, the intracellular ‘pipes’ that supply the cell with needed components. Calcium levels need to be maintained within in an extremely tight concentration range within neurons; outside of this range, reactive oxygen species are increased and there is a possibility of cell death.
Crucially, mitochondria are often closely associated with the endoplasmic reticulum, and additionally act as a temporary calcium ion store within the cell. Mitochondria may be susceptible to calcium overload that can occur due to burst firing, leading to further cell stress; taking into account the propensity for burst firing to increase calcium ion loading, and it becomes apparent that a ‘feed-forward’ mechanism of calcium overloading could create a dangerous cascade within the cell that eventually contributes to dysfunction and cell death.
That SK channels are so important in the progression of Parkinson’s disease suggests a potential target: it was shown that an SK channel facilitator can partially restore the normal electrical firing of PINK1-deficient neurons, pointing to a potential neuroprotective therapy against the early stages of Parkinson’s.
Reference:
Bishop, M. W., Chakraborty, S., Matthews, G. A., Dougalis, A., Wood, N. W., Festenstein, R., Ungless, M. A. (2010). Hyperexcitable substantia nigra dopamine neurons in PINK1- and HtrA2/Omi-deficient mice. Journal of Neurophysiology, in press. Link
